












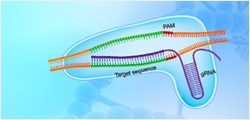































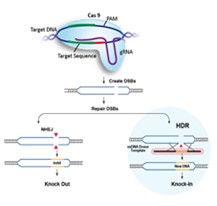
針對目標序列設計的gRNA與Cas9結合,并將Cas9引導到目標序列,目標序列的PAM序列上游3-4個堿基處,Cas9切割產生雙鏈斷裂(DSB)。
兩種方法可以修復雙鏈斷裂:1. 如果不提供修復模板或供體DNA,則會通過易出錯的非同源末端連接 (NHEJ) 進行重新鏈接,產生插入缺失從而敲除蛋白質。2. 如果提供修復模板或供體DNA,則會通過同源重組(HDR)方式修復雙鏈斷裂,以準確敲入目標基因。

資源 » 學習中心 » 常見問題解答/FAQ » 基因編輯FAQ
針對目標序列設計的gRNA與Cas9結合,并將Cas9引導到目標序列,目標序列的PAM序列上游3-4個堿基處,Cas9切割產生雙鏈斷裂(DSB)。
兩種方法可以修復雙鏈斷裂:1. 如果不提供修復模板或供體DNA,則會通過易出錯的非同源末端連接 (NHEJ) 進行重新鏈接,產生插入缺失從而敲除蛋白質。2. 如果提供修復模板或供體DNA,則會通過同源重組(HDR)方式修復雙鏈斷裂,以準確敲入目標基因。
易轉染的細胞系可以選擇質粒,轉染效率低的細胞系建議選擇病毒,期望達到編輯效率更高、毒性更低的效果可以選擇RNP(即sgRNA+Cas9蛋白)的遞送系統。
All-in-one載體系統有兩個主要優點:
雙載體中,Cas9和gRNA在不同的載體上獨立表達。如果您計劃表達多個gRNA以進行多重靶向,雙載體會更合適。對于這些應用,Cas9應首先在細胞系中穩定表達,然后可以用不同的gRNA載體轉染細胞以構建細胞庫。
CRISPR 基因編輯的載體選擇應考慮應用和細胞類型。
直接使用Cas9蛋白或RNP的優勢有:
化學合成sgRNA主要有以下優勢:
使用sgRNA時,無需在使用前對crRNA和tracrRNA雙鏈體進行退火。更重要的是,幾項研究表明,當與Cas9共同作用時,長單鏈sgRNA比crRNA:tracrRNA具有更好的穩定性,從而有更高的編輯效率1, 2。
設計您的gRNA序列包括4個步驟:
通過提供脫靶評分和染色體位置,金斯瑞的gRNA數據庫和在線設計工具將消除您在選擇gRNA序列時的疑慮,建議使用3個gRNA序列,以確保敲除和實驗準確性。
這取決于實驗目的和宿主細胞類型。與雙鏈DNA供體相比,單鏈DNA表現出顯著提高編輯效率和特異性,以及減少脫靶整合的優勢,尤其是在編輯原代細胞、干細胞和開發轉基因動物模型方面。
| 質粒DNA | dsDNA | ssDNA | |
|---|---|---|---|
| 敲入效率 | 中等 | 高 | 高 |
| 脫靶效應 | 中等 | 高 | 低 |
| 細胞毒性 | 高 | 高 | 低 |
| 成本 | 低 | 中等 | 較高 |
在單鏈DNA的生產過程中,我們進行了兩輪測序,以保證序列的準確性。我們首先通過測序選擇經過序列驗證的質粒DNA模板,以確保最終單鏈DNA產品的純度和序列準確性。此外,我們對最終的單鏈DNA產品使用直接測序來確認單鏈DNA產品的序列正確性。最終的單鏈DNA產品僅包含全長、經過序列驗證的單鏈DNA分子。通過使用我們專有的、正在申請專利的酶合成方法,金斯瑞提供的單鏈DNA產品不含雙鏈DNA。
影響CRISPR靶向效率和特異性的因素有:
在金斯瑞訂購 gRNA質粒,我們會提供一個經過序列驗證的質粒,其中包含gRNA表達和基因組結合所需的所有元素:U6啟動子、間隔(目標)序列、gRNA骨架和終止子。我們保證提供的gRNA克隆序列準確;然而,鑒于構建基因組編輯細胞系以及轉染和實驗的復雜性,我們無法保證使用我們的gRNA進行實驗的結果。如果您希望使用CRISPR技術創建經過序列驗證的KO或KI細胞系,請參閱GenCRISPR?基因編輯細胞系服務。
降低脫靶效應的方法:
基因編輯的脫靶率檢測一般有兩種方法:
一般來說主要通過以下方法:
對于點突變,建議使用非對稱單鏈DNA設計。對于大片段基因插入,有報道顯示插入基因側翼長度可從300到1000bp。在大多數情況下,500bp長度的同源臂就可以了。需要注意的是設計需要包含沉默突變的ssDNA模板,使sgRNA PAM序列發生突變,以避免二次切割。KI供體設計可能很復雜。強烈建議使用軟件(例如 snapgene)來查看和編輯序列。您可以閱讀以下文章以獲取更多設計技巧。
Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology.
